Researchers from ICVS, in collaboration with i3S, University of Coimbra and international partners, have unveiled a promising new strategy for the treatment of Machado–Joseph disease, also known as Spinocerebellar Ataxia Type 3 (SCA3). The study, published in the prestigious journal Advanced Science, focuses on preventing the aggregation of mutant ataxin-3 protein, the molecular cause of this rare, inherited neurodegenerative disease, for which there is currently no cure.
Machado–Joseph disease is caused by an abnormal expansion of a repetitive sequence in the gene encoding the ataxin-3 protein, resulting in a protein with an increased tendency to aggregate and accumulate in neurons, exerting toxic effects. Aggregates of this protein are found in the brain and spinal cord of patients. In this study, conducted with the collaboration of researchers from several institutes, the molecule CLR01 was identified as an effective inhibitor of this toxic aggregation of ataxin-3.
This molecule acts through a mechanism that inhibits protein aggregation while preserving its function. By binding to specific regions of the protein, it stabilizes ataxin-3 and prevents it from adopting multiple different conformations. In this way, it limits the exposure of regions that are more prone to aggregation, reducing the frequency with which aggregation occurs. This delays the formation of small fibers known as amyloids, a toxic form of protein aggregation, and reduces the rate of secondary nucleation, the process that enables the rapid proliferation of ataxin-3 aggregates.
To assess the beneficial effects of this molecule, several models were used. In neuronal cultures, treatment with CLR01 reversed synapse loss (the contact points between neurons responsible for communication) typically induced by the disease-associated form of ataxin-3. In animal models, the molecule improved locomotor function in the nematode Caenorhabditis elegans expressing mutant ataxin-3. In mammals, in a mouse model expressing the same protein and displaying symptoms similar to those observed in humans, treatment delayed the onset of motor symptoms and protected spinal cord neurons from the degeneration typically seen in the disease.
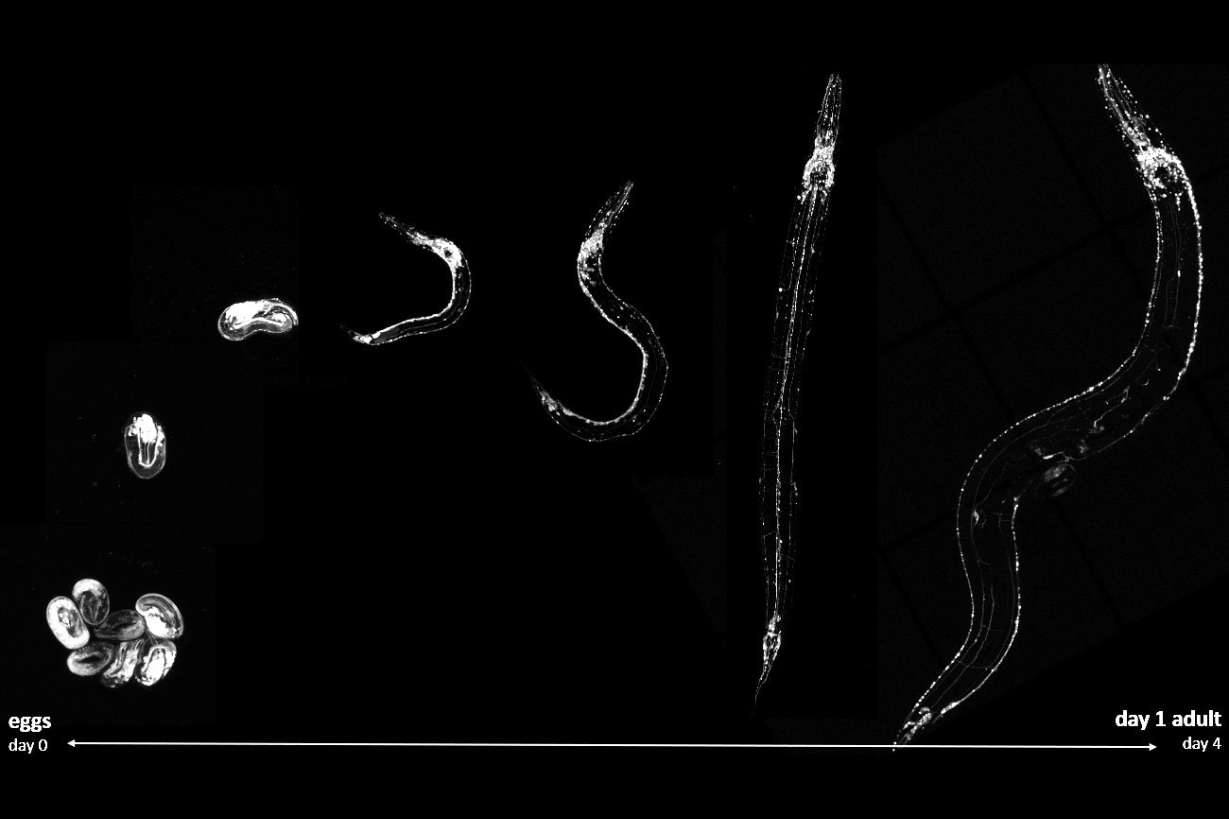
Figure 1 | Aggregation of mutant ataxin-3 protein across the developmental stages of the C. elegans model of Machado–Joseph disease.
This study, co-first authored by Sara Silva and co-led by Patrícia Maciel from TransNeu research team, also provided a significant advance in understanding the mechanisms underlying ataxin-3 aggregation toxicity. In the mouse model, a delay in disease onset was observed without a reduction in the large ataxin-3 aggregates visible under the microscope, suggesting that these large aggregates may not be the most relevant drivers of disease progression. Indeed, complementary methods showed that CLR01 alters the aggregation dynamics of the protein, promoting the formation of different, spherical aggregates with reduced toxicity. This finding suggests that the most toxic forms of ataxin-3 are irregularly shaped aggregates of varying sizes, whose formation is inhibited by CLR01.

From left to right: Sara Silva and Patrícia Maciel.
The results of this research are highly significant, as they provide mechanistic insights into ataxin-3 aggregation and validate the concept of targeting pathological aggregation as a therapeutic strategy. Moreover, identifying the binding site of this molecule on ataxin-3 paves the way for the development of new, more effective therapies capable of precisely targeting Machado–Joseph disease. This approach offers renewed hope for patients affected by this rare but highly debilitating condition, as well as for their families.




